Ancestral inference for population genetic analysis
Although identifying variable nucleotides is straightforward, inferring the direction of mutations (ancestral and derived states) can be unreliable, even among closely related sequences. Such information can greatly enhance evolutionary genome analysis by allowing prediction of fitness effects and elevating the power of statistical analyses.
We conducted computer simulation analysis to evaluate the accuracy of ancestral inference methods with a focus on detecting weak selection on base composition. Our analysis show that method assumptions can lead to substantial estimation bias. To resolve this problem, we implemented a new substitution model (GTR-NHb), which assigns a parameter-rich general time reversible (GTR) substitution model with independent parameter values for each of the analyzed branches (Matsumoto et al. 2015).
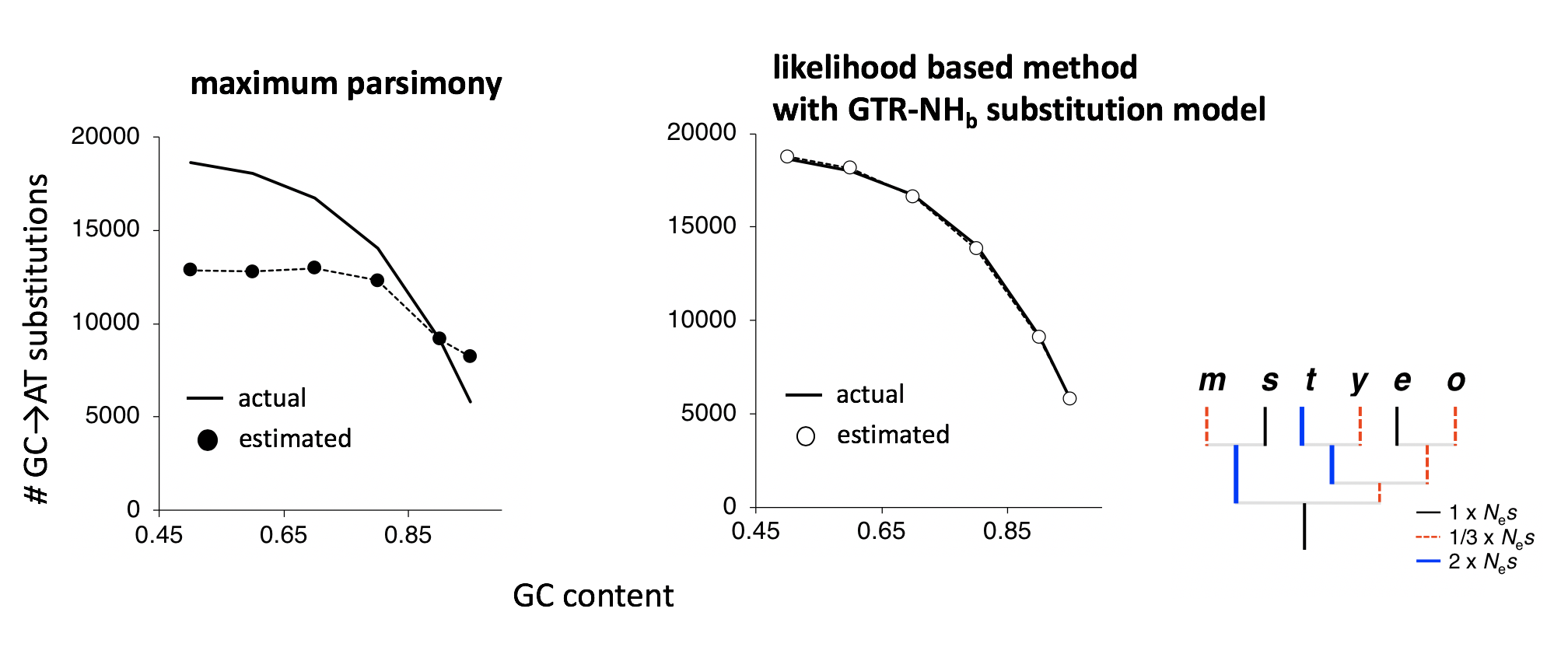
Figure 1. Actual vs estimated numbers of inferred substitutions: parsimony vs maximum likelihood.
Data were simulated for a gene tree similar to the Drosophila melanogaster subgroup (shown on the right) under weak selection favoring G/C where selection intensity varies among lineage. Lineages with elevated selection intensity are shown in blue and those with decreased selection intensity in red. Maximum parsimony (left) and likelihood based method with GTR-NHb model (right) were used for the ancestral inference. Data are shown for the m lineage in both graphs.
We also developed a new method (bifurcating tree with weighting, BTW), in which likelihood based probabilities of ancestral states are weighted by population genetics expectations for the frequencies of polymorphic mutations (Matsumoto and Akashi 2018). Our computer simulation analysis shows that BTW can substantially improve the accuracy of ancestral inference for polymorphic mutations in recombining genomes. Using this method, we can estimate unfolded site frequency spectrum to detect weak selection.
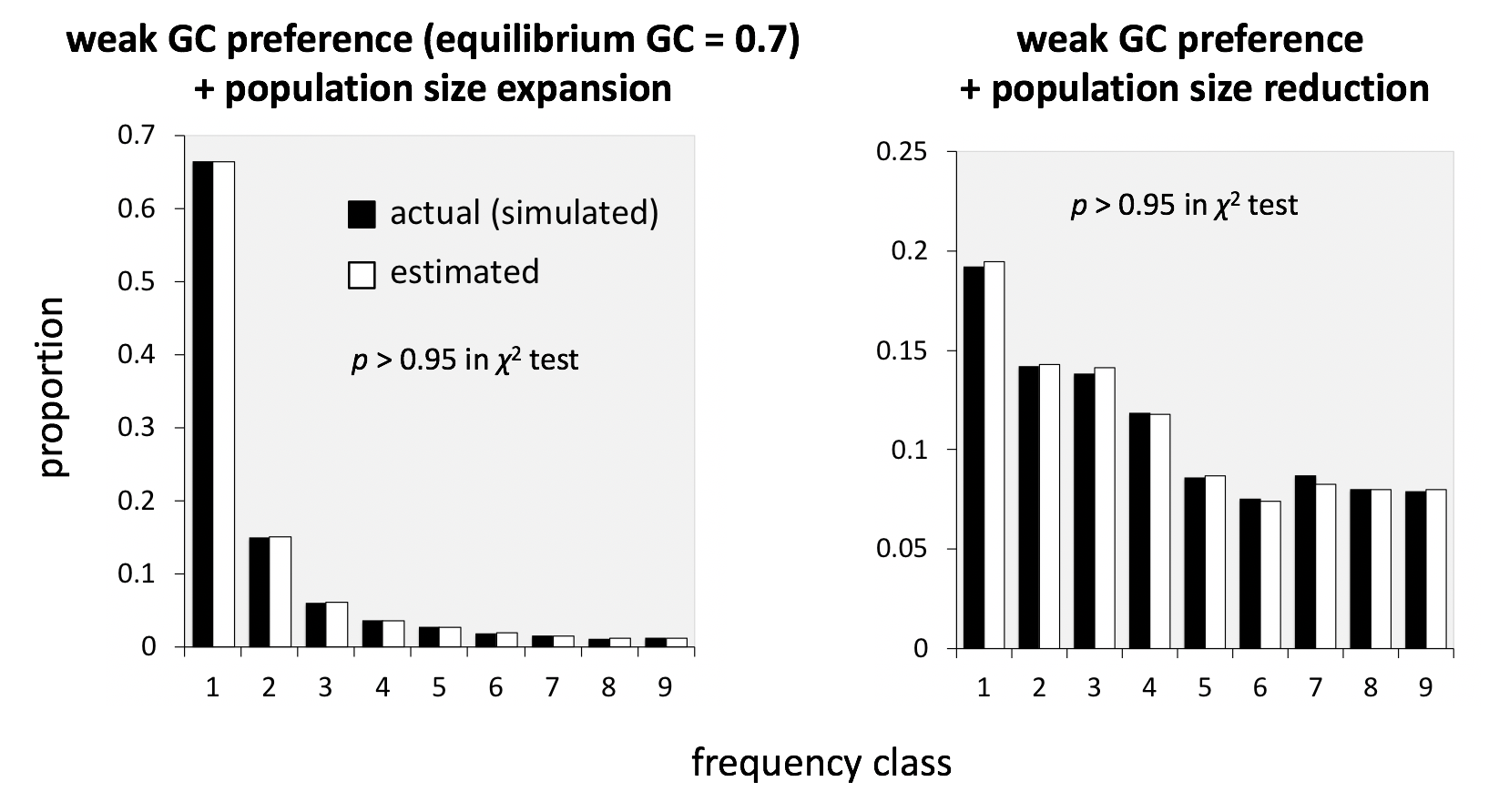
Figure 2: Actual vs estimated numbers of inferred substitutions: weighting using population genetic expectations.
Data are shown for BTW analysis of a data set of 10 samples (alleles). Results in two evolutionary scenarios that show contrasting site frequency spectra (uSFS) are shown. For both scenarios, BTW showed accurate estimation (high p values of chi-square goodness of fit tests).
References
- Ancestral inference and the study of codon bias evolution: implications for molecular evolutionary analyses of the Drosophila melanogaster subgroup
- PLoS ONE 2:e1065. 2007.
- Evaluation of Ancestral Sequence Reconstruction Methods to Infer Nonstationary Patterns of Nucleotide Substitution
- Genetics 200:873–890. 2015.
- Distinguishing Among Evolutionary Forces Acting on Genome-Wide Base Composition: Computer Simulation Analysis of Approximate Methods for Inferring Site Frequency Spectra of Derived Mutations
- G3 8:1755-1769. 2018.